生命所必需的每一次基础生物学进展几乎都是由蛋白质带来的。蛋白质参与创建细胞和组织并保持着它们的形状;构成维持生命所需化学反应的催化酶;充当分子工厂、转运工具和马达;充当细胞通讯的信号和接收器等等。
蛋白质由很多氨基酸长链组成,通过折叠成精确的 3D 结构来完成无数的任务,这些结构控制着它们与其它分子互动的方式。蛋白质的形状决定了其功能以及它在疾病中的功能紊乱程度。阐明蛋白质的结构是所有分子生物学的核心,更是治疗患者、拯救生命、改变生活的医学发展的核心。
近年来,根据氨基酸序列预测蛋白质折叠结构方面的计算方法已经取得了很大进展。如果能够充分实现,这些方法可能会改变生物医学研究的方方面面。然而,现在的方法在可测定的蛋白质的大小和范围上是有限的。
最近,哈佛大学医学院 Blavatnik 研究所系统生物学家 Mohammed AlQuraishi 发布了一项新的研究,他根据氨基酸序列,利用深度学习实现了任意蛋白质 3D 结构的高效预测。在 4 月 17 日的 Cell Systems 期刊中,Mohammed AlQuraishi 详细介绍了这种通过计算确定蛋白质结构的新方法,利用该方法实现的准确率可媲美当前最佳方案,但速度提高了 100 万倍。
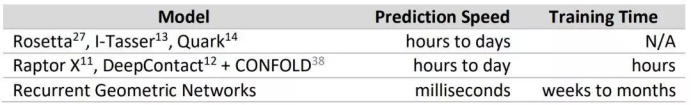
如上所示,AlQuraishi 提出的循环几何网络在预测速度上快了 6 到 7 个数量级。其中上表第一行是目前已经建立起来的复杂方法,这些方法严重依赖于模拟和采样。第二行对应于协同演化(co-evolution)的方法,它也会有一个学习过程。最后一行就是作者提出的一种端到端的可微分方法。
说起来简单
虽然成功率高,但利用物理工具来鉴别蛋白质结构的过程既昂贵又耗时,即使是使用现代技术(如低温电子显微镜)同样如此。因此,绝大多数蛋白质结构以及致病突变对这些结构的影响目前仍是未知的。
蛋白质折叠方式的计算方法有可能大大降低确定蛋白质结构的成本和时间。但经过近四十年的不懈努力,这个难题仍未解决。
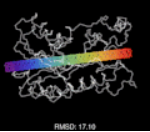
可视化模拟 AlQuraishi 的蛋白质折叠深度学习方法。模型通过反复预测某个结构(彩色)并将其预测与真实结构(灰色)对比来实现自训练。对数千种已知蛋白质重复此步骤,模型在每次迭代中都会学习并提高自己的准确率。
做起来难
为了应对这一挑战,科学家利用氨基酸的特点,即它会在物理法则的约束下相互作用,从而寻找更具潜力的状态。目前最先进的算法通过超级计算机或众包计算来寻找蛋白质结构,它们基本上都是在模拟极其复杂的氨基酸物理相互作用。为了降低这些方法对大规模计算的需求,它们依赖于将新序列映射到预定义的模板,且这些蛋白质结构模板都是之前通过实验确定的。
其它如DeepMind的AlphaFold则采用了另一种方法,它利用深度学习进一步预测蛋白质结构。这类方法会分析大量的基因数据,其内在包含了蛋白质的设计蓝图,目前受到了极大的关注。
AlphaFold是之前非常受关注的一项研究,它利用神经网络来预测氨基酸对之间的距离,以及连接它们的化学键之间的角度。 AlphaFold参加了 CASP 竞赛,它首次参赛就在 98 名参赛者中名列榜首,准确地从 43 种蛋白质中预测出了 25 种蛋白质的结构,而同组比赛中获得第二名的参赛者仅准确预测出了 3 种。
然而,这些 AI 方法并不能仅基于蛋白质的氨基酸序列预测结构。这一点会限制AlphaFold等方法的能力,因为在没有先验知识的情况下它们很难决定蛋白质结构,其在演化独特的蛋白质或手工设计新蛋白质上能力有限。
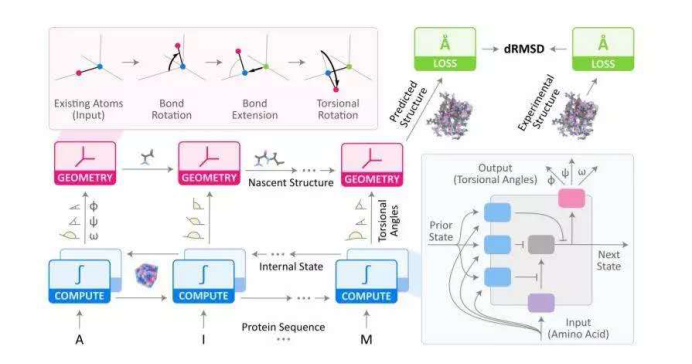
针对这些问题,AlQuraishi 主要从四个核心概念出发构建新的解决方案。首先我们应该要使用循环神经网络编码蛋白质序列;其次通过扭转角度参数化局部蛋白质的结构,从而允许模型在不破坏共价化学性质的情况下对各种结构进行推理;再者,我们还应该通过循环几何单元耦合局部蛋白质结构和它的全局表征;最后,使用一种可微损失函数来捕捉预测结构与实际结构之间的差别。

新的前景
这种极速使得蛋白质预测变得更加简单,以前无法想象的应用也都会慢慢冒出来。AlQuraishi 表示:「新模型还没有立即用于新药开发或设计,因为它的准确度大约在 6 埃(一亿分之一厘米)左右,与解决蛋白质原子结构所需的 1 到 2 埃还有一定距离。」
但是在深度学习快速发展的今天,这种方法将越来越强大,例如如何进一步整合化学和物理知识就值得我们认真探讨。也许以后的新药开发成本会越来越低,医疗费用也会越来越低。
转自:机器之心
